| Dictionnaire médical |
Abréviations M Macr Macro Macu Magn Mal Malacie Maladie Maladie de Malaria Malt Mamm Mamm(o)- Man Manœuvre de Mart Maso Mass Mast Mast(o) Maïe Melæ Mens Menstr Meur Micr Micro Mict Miction Migr Migraine Mili Minéral Miop Mito Mnèm Mném Mnés Moel Mole Moll Mona Mono Morb Morp Mort Moru Mour Muc Muci Muco Mult Musc Muscle Myas Myco Mycé Mydr Myo Myri Myring Myth Myx Myél Méat Méca Méde Médi Médiastin Médu Médull Méga Méio Méla Mélan Méni Méning Ménisc Méno Mér Méro Més Méso Méta Méth Méthode de Métr Métro Mété Météor Môla Môle
| MA | |
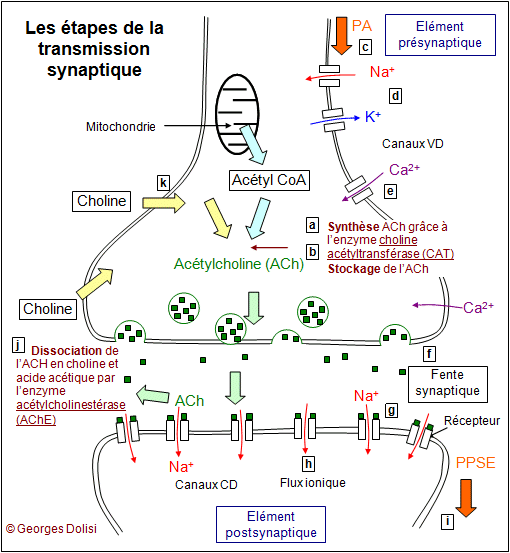
| Neurologie, psychiatrie psychologie Abrév. Maladie d'Alzheimer. * Alzheimer : du neuropsychiatre allemand Aloïs ALZHEIMER (Markbreit, 1864 - Breslau, 1915). Le nom d'Aloïs Alzheimer est lié à la maladie dont il décrivit pour la première fois les symptômes en 1906, lors de la 37e Conférence des psychiatres allemands. C'est le professeur Emil Kraepelin qui proposa par la suite de désigner ce type de démence par le nom de son collègue. Né le 14 juin 1864, dans la petite ville bavaroise de Markbreit, au sud de l'Allemagne, Alzheimer suit de brillantes études de médecine à Berlin, Würzburg et Tubïngen, dont il sort licencié en 1888. La même année, il commence à travailler à l'hôpital psychiatrique de Francfort. En 1906, à la mort d'une ancienne patiente qu'il avait suivie, Alzheimer pratique l'autopsie de son cerveau. Utilisant la technique histologique d'imprégnation argentique, il met en évidence les trois types de lésions cérébrales caractéristiques de la maladie qui fera sa renommée : plaques séniles, dégénérescence neurofibrillaire et atrophie cérébrale. La maladie d'Alzheimer (MA) est la plus commune des démences séniles et les formes purement héréditaires représenteraient moins de 0,5% des malades, alors que plus de 90% sont des formes sporadiques. En France, en 2010, 860 000 personnes, environ 1 homme sur 8 et 1 femme sur 4 - sont touchées (28 millions dans le monde) et on recense 90 000 nouveaux cas par an. Cette pathologie est, en l'état actuel des connaissances, irréversible, car les médicaments prescrits ne peuvent qu'en ralentir l'évolution. Voici les 10 signes les plus évidents, édités par l'Association France Alzheimer ( http://www.francealzheimer.org ) : 1 - Des pertes de mémoire, notamment à propos d'événements ou d'informations récents et les mêmes questions répétées à intervalles brefs. 2 - Difficultés à accomplir des tâches quotidiennes : un malade peut préparer correctement un repas et oublier de le servir. 3 - Des problèmes de langage : le malade oublie des mots simples et les remplace par d'autres qui sont totalement inappropriés. 4 - Désorientation dans le temps et dans l'espace : les personnes malades perdent progressivement le sens de l'orientation et se perdent facilement, même à faible distance de leur habitation. 5 - Pertes du jugement : le malade fait subitement abstraction du monde extérieur, prend des risques inutiles, oublie ses rendez-vous. 6 - Raisonnements abstraits : toutes les formalités administratives concernant la banque, la santé, les impôts ... deviennent un véritable calvaire, voire impossibles. 7 - Perte d'objets : les objets de la vie courante ou personnels sont placés dans des endroits insolites (trousseau de clés dans le réfrigérateur, montre dans le four par exemple) et ne sont retrouvés que par hasard. 8 - Troubles du comportement : l'humeur du malade est susceptible de subir des changements brutaux, passant sans raison de la douceur à l'agressivité. 9 - Changement de personnalité : les changements de la personnalité peuvent affecter un malade au point de le rendre méconnaissable même pour ses proches parents. 10 - Pertes d'initiative : le malade n'a plus de motivation, plus goût à rien, même pour ce qui étaient ses activités préférées. Ces symptômes sont parfois associés à une incontinence, une agitation. On sait aujourd'hui que, dans 80 % des cas, ces patients présentent une destruction localisée de cellules cérébrales particulières et une disparition des récepteurs nicotiniques à l'acétylcholine. Actuellement, on en est encore au stade des hypothèses : - neurochimique : déficit local (cortex et hippocampe) de l'enzyme choline acétyl transférase (ou acétylcholinestérase) provoquant une diminution de l'acétylcholine, neurotransmetteur indispensable pour les neurones de ces zones ; - une hypothèse génétique repose sur l'existence d'antécédents familiaux chez 15% de malades. La transmission se fait selon le mode autosomique (chromosome non sexuel) dominant (un seul parent atteint et l'enfant est atteint). Les gènes responsables, codant la protéine bêta-amyloïde, seraient situés sur les chromosomes 21, 19 et 14. - Hypothèse virale : le virus aurait besoin d'un certain contexte immunitaire et toxique pour s'exprimer ; - hypothèse des radicaux libres, qui ont un effet hautement destructeur et provoqueraient des dégâts sur le tissu nerveux. * Les plaques séniles ou plaques bêta amyloïdes sont formées essentiellement par l'accumulation de la protéine anormale Aß, elle-même résultant d'une scission de la protéine codée par le gène APP. * DNF ou dégénérescence neurofibrillaire : enchevêtrement anormal des fibres nerveuses, à cause de la présence d'une protéine Tau anormale, généralement associée aux microtubules du cytosquelette et qui va progressivement provoquer la mort des neurones. Cette protéine anormale est dite hyperphosphorylée, car elle subit jusqu'à 9 phosphorylations chez les patients atteints de MA, au lieu de 2 à 3 phosphorylations normales. C'est une protéine qui est normalement associée aux microtubules du cytosquelette, et Il en résulte logiquement des microtubules totalement enchevêtrés et qui vont gêner le transport axonal. Exemple : le transport de l'APP ou précurseur de la protéine amyloïde - Amyloid Protein Precursor. Elle est véhiculée dans l'axone vers l'extrémité nerveuse et la synapse où elle joue un rôle important dans la plasticité neuronale et la neurotransmission. Il faut noter que cette APP est normalement très répandue dans l'organisme jouant plusieurs rôles au niveau des cellules et en particulier dans le tissu nerveux : c'est une molécule qui va permettre des interactions entre cellules, c'est aussi un récepteur de surface et un facteur de croissance qui va intervenir dans la formation du cytosquelette, le régulation du calcium intracellulaire, le formation des synapses, la modulation des cholinestérases entre autres. On trouve deux isomorphes anormaux de l'APP (en plus de la molécule normale) chez les malades d'Alzheimer. * L'atrophie corticale, notamment au niveau de l'hippocampe et des zones voisines. Cette atrophie cérébrale ou perte de matière cérébrale peut aller jusqu'à 7 à 8% en 10 ans. Le volume de la boîte crânienne étant constant, cette perte est compensée essentiellement par une augmentation des ventricules du cerveau (cavités remplies de LCR ou liquide céphalorachidien) et par un élargissement du sillon interhémisphérique et des sillons secondaires. Cette perte de matière est particulièrement visible au niveau des zones riches en fibres nerveuses cholinergiques, c'est-à-dire des neurones qui utilisent, au niveau de leurs synapses, l'acétylcholine comme neurotransmetteur (NT) et donc des récepteurs nicotiniques et/ou muscariniques. Et c'est là tout le problème, car la protéine bêta-amyloïde est aujourd'hui considérée comme le plus puissant neuromodulateur (= un neuromodulateur est une substance qui n'agit pas directement sur les PPSE ou PPSI comme un NT par exemple, mais qui va agir de façon indirecte) cholinergique (= qui va agir sur les neurones dont le NT synaptique est l'ACH) négatif (= qui va agir négativement sur ces synapses en ralentissant leur fonctionnement) connu. Cela signifie que, avant même son dépôt sous forme de plaques séniles, cette protéine bêta-amyloïde va inhiber le fonctionnement de ces synapses, en agissant sur le canal de recapture de la choline (ou ensemble des réactions de récupération de l'ACh qui a servi dans la synapse et remise à disposition dans les vésicules de l'élément présynaptique). A noter que c'est probablement la structure en "feuillet bêta" de cette protéine qui lui confère son caractère d'insolubilité et sa haute toxicité. (Pour en savoir plus sur la structure des protéines, voir la page "proto, protéino", lettre "P"). Commentaire du schéma "La synapse cholinergique" : Rappel : fonctionnement normal d'une synapse cholinergique : La synapse cholinergique Dans le neurone présynaptique, la synthèse du neurotransmetteur (dans notre exemple l'acétylcholine ou ACh) est faite à partir de la choline et d'acétyl-coenzyme A (ou acétyl-CoA) , présent dans les mitochondries et résultant de la dégradation du glucose en pyruvate puis en ACh. Cette synthèse est possible grâce à la présence d'une enzyme CAT (choline acétyltransférase). Le neurotransmetteur est ensuite stocké dans des vésicules de transport qui l'amèneront près de la membrane, côté fente synaptique. Il y restera tant qu'aucun PA (potentiel d'action) n'arrivera. Rappelons simplement que le potentiel de repos d'un neurone est de -70 mV. Un PA est une onde de dépolarisation provoquée par des mouvements ioniques (entrée et sortie d'ions). Cette étape représente l'arrivée d'un PA et les mouvements des ions à travers des protéines canaux très spécifiques : les canaux voltage-dépendants. C'est l'ouverture progressive de ces canaux transmembranaires qui permet la propagation des PA Les différentes phases du potentiel d'action : L'ouverture des canaux Na+-VD permet une entrée massive d'ions Na+ car la concentration extracellulaire de ces ions est naturellement supérieure à la concentration intracellulaire. Cette entrée se fait donc par diffusion simple, en fonction du gradient des concentrations et ne consomme pas d'énergie. Il en résulte une inversion du potentiel cellulaire (dépolarisation) qui passe ainsi de - 70 mV à + 40 mV. Ces canaux se ferment très rapidement et le flux d'ions Na+ cesse. En même temps, les canaux K+-VD s'ouvrent et cette fois, c'est un flux sortant d'ions K+ qui se produit car leur concentration intracellulaire est naturellement supérieure à la concentration extracellulaire. Il en résulte une repolarisation et un retour à la valeur de repos de - 70 mV. Comme pour les canaux Na+-VD la fermeture est quasi immédiate (après un délai toutefois un peu plus long, entraînant une légère hyperpolarisation). Le tout n'aura duré que 2 à 3 ms (millisecondes). Le déséquilibre ionique de l'intérieur de la cellule par rapport à l'extérieur, qui permet une valeur de repos de - 70 mV, est constamment entretenu par une pompe Na+ - K+ - ATPase qui provoque un mouvement d'ions inverse à celui de la diffusion provoqué par les gradients de concentration. L'arrivée d'un PA provoque l'ouverture de nouveaux canaux appelés canaux Ca++ (ou Ca2+) voltage-dépendants et l'entrée d'un flux d'ions Ca++ dans le compartiment présynaptique. La présence de calcium sous forme ionique est indispensable pour la suite des événements. C'est en effet l'intrusion des ions Ca2+ qui va permettre la fusion des vésicules de transport contenant le neurotransmetteur, avec la membrane présynaptique. Des centaines de vésicules vident ainsi leur contenu dans l'espace synaptique par exocytose. Ces molécules viennent se fixer sur les récepteurs spécifiques présents en grand nombre sur la membrane postsynaptique. Ces récepteurs sont des protéines transmembranaires qui vont s'ouvrir au moment de la fixation du neurotransmetteur. Ce sont donc des canaux ioniques chimio-dépendants (canaux CD). L'ouverture de ces canaux CD permet des mouvements ioniques qui vont induire une perturbation du potentiel de membrane postsynaptique, qui va déterminer l'état global d'excitation du compartiment postsynaptique. Il en résulte un potentiel postsynaptique qui sera renouvelé tant que des potentiels d'action parviendront à l'extrémité présynaptique. C'est donc la fréquence des potentiels présynaptiques qui détermine l'importance des molécules de neurotransmetteur dans la fente synaptique et donc la réponse du neurone postsynaptique. La synapse n'est pas une simple jonction : elle transforme une information nerveuse, codée en modulation de fréquence, en une autre information, codée en amplitude. A noter que, selon le type de récepteur et donc de neurotransmetteur, le potentiel peut être excitateur (PPSE ou potentiel postsynaptique excitateur) ou au contraire inhibiteur (PPSI ou potentiel postsynaptique inhibiteur). La présence du neurotransmetteur dans l'espace synaptique et sur les récepteurs est extrêmement fugace, car il est presque instantanément détruit par une enzyme : dans notre exemple, il s'agit de l'acétylcholinestérase, qui est décomposée en choline et en acide acétique (ou ion acétyl). Ces molécules de choline sont récupérées par le neurone présynaptique et interviennent dans la synthèse de nouvelles molécules de neurotransmetteur. On peut avoir ainsi jusqu'à 50 PA par seconde. Il s'agit là encore de réactions complexes et nombreuses connues sous le nom de "canal de recapture de la choline". Les apolipoprotéines E et la MA : les apo E sont essentiellement des transporteurs de cholestérol et de phospholipides et ils permettent de ce fait d'alimenter les cellules de l'organisme en cholestérol et en phospholipides. C'est ce qui explique qu'ils interviennent dans la restructuration ou la réparation des membranes neuronales (donc des cellules nerveuses, dans lesquelles on trouve essentiellement des phospholipides et du cholestérol). On connaît aujourd'hui 3 formes d'apo E (E2, E3 et E4), issues de 3 allèles du gène qui code l'apo E (sur le chromosome 19) : e2, e3 et e4. On a constaté (dans des études publiées en 1993) que l'allèle e4, donc l'apo E4, est environ 4 fois plus présent (20%) chez les personnes atteintes de la MA (maladie d'Alzheimer) que chez les non malades (environ 5%). Des études récentes ont montré que les porteurs de e2 ont moins de cholestérol sanguin, alors que ceux qui ont l'allèle e4 présentent une hypercholestérolémie. A signaler également le fait que les apolipoprotéines sont reconnues par des récepteurs de la membrane cellulaire, ce qui permet leur passage de part et d'autre de cette membrane. Pour simplifier : * l'allèle e4 (Apo E4) représente un réel facteur de risque pour les MCV (maladies cardiovasculaires) et la MA, même si elle est neuroprotectrice (car c'est la moins active dans cette protection, de toutes les apolipoprotéines E et les porteurs de E4 réparent moins bien leurs cellules nerveuses que les autres), car elle favorise la formation de plaques séniles en favorisant l'agrégation du peptide Aß ou peptide amyloïde bêta). * l'allèle e2 (Apo E2) par contre, représente une réelle protection contre les MCV et la MA. Les résultats sont immédiats : * la choline n'est plus correctement recapturée par le compartiment présynaptique * la synthèse d'ACh ne se fait plus suffisamment dans ce compartiment, car il y a déficit de choline, * il n'y a plus assez de vésicules d'ACh pour "alimenter" la fente synaptique * la transmission des PA (potentiels d'action) ne se fait plus correctement La conséquence est terrible et irréversible : On sait qu'il y a constamment un nombre important de fragments de membrane plasmique qui sont échangés avec des inclusions par ex., ce qui ne gène pas une cellule normale ou saine. Dans le cas de ces neurones qui sont "en manque" de choline, ils vont faire ce que les biochimistes appellent l'autophagie ou autocannibalisme : ils vont détruire des portions relativement nombreuses et importantes de leur membrane plasmique, pour en utiliser la choline des molécules de phosphatidylcholine provenant de cette membrane. En effet, chez la plupart des mammifères, quatre phospholipides principaux prédominent dans la membrane plasmique de la plupart des cellules : la phosphatidylcholine, la sphingomyéline, la phosphatidylsérine et la phosphatidyléthanolamine. Entre ces molécules se trouve un nombre plus ou moins important de molécules de cholestérol. Ces blessures sont si importantes et nombreuses que les cellules en sont irrémédiablement affaiblies, soumises en plus a l'attaque des radicaux libres émis par ces mêmes plaques séniles. Le traitement repose aujourd'hui essentiellement sur des antidépresseurs non tricycliques et des médicaments qui pallient la carence en acétylcholine (Donepezil). Des équipes de chercheurs travaillent sur une immunothérapie consistant à injecter des protéines "vaccinales" ressemblant aux protéines bêta-amyloïdes, qui s'accumulent en plaques séniles. Les résultats sur les souris sont encourageants (diminution des dépôts) et les essais chez l'homme ont commencé. La présence d'aluminium dans l'alimentation et notamment dans l'eau de boisson a également été soupçonnée d'être un facteur environnemental non négligeable, puisqu'elle multiplierait par 2 ou même 3 le risque de développer une MA. Le 26 janvier 2010, le professeur Etienne-Emile BEAULIEU et ses collaborateurs de l'INSERM (Institut National de la Santé et de la Recherche Médicale - Inserm U788 "Stéroïdes, neuroprotection et neurorégénération", université Paris XI) ont présenté, à l'Académie des Sciences, leur découverte d'une interaction entre une protéine FKBP52 et la protéine Tau responsable, par sa présence excessive, du processus de dégénérescence nerveuse. Cette protéine FKBP52 s'attaquent aux protéines Tau : "Les protéines FKBP52 diminuent la concentration et l'action de la protéine Tau, et inhibent la formation de buissons de cette protéine Tau dans les neurones." D'où un double espoir : celui de pouvoir mesurer la quantité de protéines FKBP52 chez les personnes pour évaluer leur risque ultérieur de développer une maladie d'Alzheimer (l'étude devrait bientôt commencer à l'hôpital Charles Foix d'Ivry, en région parisienne) et, surtout, celui de trouver des médicaments capables de stimuler cette protéine anti-Tau. "Les laboratoires ont déjà dans leurs tiroirs des molécules ayant ce mode d'action, mais dont ils ne savaient que faire", selon le Pr Beaulieu. Néanmoins, un autre médicament "anti-Tau", agissant de façon différente, est actuellement expérimenté chez l'homme. Le 10 février 2012, une publication dans la revue américaine Science suscite un très grand espoir. Extraits : Des souris programmées génétiquement pour développer la maladie d'Alzheimer, ont été traitées par des scientifiques américains avec un médicament aticancéreux : le Bexarotene. En 72 heures, ce médicament a non seulement fait disparaître plus de 50% des plaques bêta-amyloïdes, mais elles ont retrouvé la mémoire et le sens de l'odorat. Ces commentaires ont été faits par le Dr. Daniel WESSON, professeur adjoint de neurosciences à la faculté de médecine Case Western à Cleveland, dans l'Ohio, principal co-auteur de cette étude et de cette publication. A noter que chez les humains, la perte de l'odorat est souvent l'un des premiers signes de la maladie d'Alzheimer. Paige CRAMER, un autre chercheur de la faculté de médecine Case Western a déclaré cette avancée "Sans précédent" et le Dr. Gary LANDRETH, professeur de neurosciences dans la même faculté et principal auteur de l'étude : "Nous sommes encore au tout premier stade de nos efforts pour transformer cette découverte de recherche fondamentale en un traitement". Les plaques bête-amyloïdes se forment naturellement dans le cerveau mais le système immunitaire, en vieillissant, perd la capacité de les éliminer. Il apparaît que le Bexarotène reprogramme les cellules immunitaires dans le cerveau pour qu'elles puissent de nouveau "dévorer" les dépôts amyloïdes.
© Georges Dolisi | |
| < vers le menu principal | Politique de confidentialité | App Privacy Policy | Contact | ^ haut de la page |
| © 2006, 2020 Medicopedia | Powered by JIMaroc |